The blog post titled ‘The genetic lottery’ came about as a result of my attempt to understand the link between Ruzbeh’s mutation and the severity of his symptoms. His mutation being a missense mutation (one where just one amino acid in the protein coded for by the gene, gets replaced by another), is generally considered less harmful/severe than let’s say, a deletion. At least by laypeople, which is what I am.
Now here is some feedback from a regular reader (my dear husband) about that post:
I didn’t understand a thing!
What on earth was that all about?
That was not a blog post; that was a white paper on Norrie disease!
Right… Um…. Well… Yes…. :-))))
Yet another reader (from the Norrie support group) was left feeling confused, not only with respect to the contents of the post, but also its title.
So here is another shot at it – simpler, shorter, and trying to address the above mentioned concerns.
For fear of writing yet another mile long post, I am going to split it into two – an explanation of the title in today’s post, and that of the content in the sequel to this post.
So here we go again.
(a) My use of the phrase ‘genetic lottery’ referred to the fact that some of those born with Norrie Disease (ND) have it worse than others. These are the 30-50% of cases who manifest neurological problems in addition to the other symptoms of the disorder. A double whammy, if you like.
We might well ask, why only 30-50%, why not 100%? There is, as yet, no answer to this question. My hypothesis is, that this has to do with the severity of the mutation. Not that I’m an expert of anything; I’m not. Most scientists on the other hand, tend to point the finger to ‘other genes’. Since the functioning of the brain is a puzzle yet to be solved, we could attribute the mental deficit associated with ND to ‘other genes’. I’m just not sure how we could explain the high figure of 30-50% when the incidence of autism in the general population is pegged at between 1% and 2% (depending on which report you happen to be reading).
While it is my belief that there is a definite link between severity of mutation and severity of symptoms, one must keep one’s mind open to the possibility of modifier genes making the outcome of an already severe mutation even worse. As the name suggests, modifier genes are genes which modify the clinical outcome of another gene. Click here to see how modifier genes modify the outcome of mutations of a gene called CFTR. Research is needed to identify the modifier genes for ND.
So, if we were to suppose that there is a link between the severity of the mutation and the severity of symptoms, then it begs the question - which mutations of NDP (the Norrie disease gene) are more severe than others? This is exactly the question I was trying to address in ‘The genetic lottery’, and I hope to explain it more clearly in the sequel to this post.
(b) Another meaning of the phrase ‘genetic lottery’ could have been (but in my blog post, was not), the fact that some people are born with ND at all. It is after all, a rare disease. Just how rare? There is no official estimate of the number of cases worldwide. The orphanet description for ND mentions that there are 400 published cases to date, which is neither here nor there. It tells us nothing about the incidence or the prevalence of the disorder.
(c) Yet another meaning of the phrase ‘genetic lottery’ could have been (but in my blog post, was not) the fact that some disease causing mutations of the gene NDP do not result in ND. Instead they might lead to such lovelies as Coat’s disease, ROP (retinopathy of prematurity) stages 4b & 5, FEVR (familial exudative vitreoretinopathy), and PHPV (persistent hyperplastic primary vitreous). None of these other diseases is as severe as ND. An estimated 77% of mutations of NDP result in ND.
Here we might well ask – how on earth is it possible that mutations of one gene result in several diseases? While searching for an answer to this question, I came across an article which explains how different mutations of a gene called SHANK1 lead to two distinct disorders – autism in one case and schizophrenia in the other. A study was conducted to try and find the reason why different mutations of SHANK1 resulted in two distinct psychiatric disorders. The result of this study? The mutation which led to autism (deemed to be the more severe of the two disorders) resulted in a complete loss of gene product (i.e. the protein coded for by the gene) whereas the mutation which caused schizophrenia resulted in a truncated product (the resulting protein was about half the size). In other words, the severe mutation led to a severe outcome whereas a less severe mutation led to a less severe outcome. Might this explain why different mutations of the gene NDP result in different diseases? Perhaps. It could also be the case that in addition to the type of mutation, modifier genes (see (a) above) also contribute to mutations of the same gene resulting in different diseases.
(d) And finally, here is yet another interpretation of the phrase ‘genetic lottery’. There are cases where the same mutation of the gene NDP has resulted in different outcomes/symptoms. At times this has been the case even within the same family. According to scientists, the answer lies in – you guessed it - modifier genes (see (a) above).
I hope things are a lot clearer this time round, and I’m off to write part II of this post.
Till next time then,
Meenu.
Wednesday, September 13, 2017
Thursday, June 1, 2017
Of letters and fact sheets
This is going back quite a while, but in a previous post I had spoken about a letter which I was going to send to the National Eye Institute (NEI). The idea was to request the NEI to convene a meeting of researchers dealing with various aspects of retina related research, in order to ascertain what could be done to reverse the blindness caused by Norrie Disease (ND).
But then stuff happened, life happened, Ruzbeh’s epileptic attacks happened, and the letter got relegated to the background.
And as is often the case with the passage of time, ideas change, more research takes place, and plans get revised.
As I pick up the thread and continue on my journey, I’m more inclined to reach out to individual researchers, just as I have been doing for other symptoms. It will be interesting to see if I can engage with anyone at all - try talking to folks about a cure for ND related blindness and they invariably steer the discussion to vision substitution systems.
Speaking of the NEI, here is a little anecdote:
A long, long time ago - like maybe a year ago – I happened to be poking around the NEI’s website, and I came across a tab labelled “A-Z Diseases and Disorders ”. My immediate thought was ooooh, let’s see what it says for Norrie disease. Surprise, surprise, there was no mention of ND. Well not under ‘N’ anyway. So I poked around a bit more and listed under ‘R’ was “Rare Diseases”. Okaaay, I thought, it’s got to be here. Bigger surprise – no mention of ND there either.
I’m sat there thinking, here we are, living with the crowning glory of eye diseases. We ought to have made it to the top of the list. LOL.
So I wrote to the NEI, as one does, to enquire why their website does not feature ND anywhere. I received a reply which said “currently, the NEI does not have a fact sheet on this condition which is why it is not listed under “Rare Diseases”.” It went on to list other websites where ND related information could be obtained. You know, the usual stuff – NORD, OMIM, NDA, PubMed. Actually I could even add my own blog to this list. But I digress.
Now I’m sat there scratching my head - fact sheet? What is a fact sheet? And whatever it is, why can't we make one for ND?
So I wrote back. What is a fact sheet? What are the criteria which determine which diseases get to have a fact sheet? What can we, the Norrie community, do in order to have a fact sheet about ND? Etc.
This time the line went dead. No reply.
Silly me with my silly questions.
Do you know why there is no mention of Norrie disease on the NEI website? If so, I would love to hear from you. As always, I can be reached at maladiedenorrie@gmail.com.
Till next time then,
Meenu.
PS: The aforementioned tab on “A-Z Diseases and Disorders” appears to have been replaced by “Search A-Z” on the NEI website (just below the search box on the top right corner).
But then stuff happened, life happened, Ruzbeh’s epileptic attacks happened, and the letter got relegated to the background.
And as is often the case with the passage of time, ideas change, more research takes place, and plans get revised.
As I pick up the thread and continue on my journey, I’m more inclined to reach out to individual researchers, just as I have been doing for other symptoms. It will be interesting to see if I can engage with anyone at all - try talking to folks about a cure for ND related blindness and they invariably steer the discussion to vision substitution systems.
Speaking of the NEI, here is a little anecdote:
A long, long time ago - like maybe a year ago – I happened to be poking around the NEI’s website, and I came across a tab labelled “A-Z Diseases and Disorders ”. My immediate thought was ooooh, let’s see what it says for Norrie disease. Surprise, surprise, there was no mention of ND. Well not under ‘N’ anyway. So I poked around a bit more and listed under ‘R’ was “Rare Diseases”. Okaaay, I thought, it’s got to be here. Bigger surprise – no mention of ND there either.
I’m sat there thinking, here we are, living with the crowning glory of eye diseases. We ought to have made it to the top of the list. LOL.
So I wrote to the NEI, as one does, to enquire why their website does not feature ND anywhere. I received a reply which said “currently, the NEI does not have a fact sheet on this condition which is why it is not listed under “Rare Diseases”.” It went on to list other websites where ND related information could be obtained. You know, the usual stuff – NORD, OMIM, NDA, PubMed. Actually I could even add my own blog to this list. But I digress.
Now I’m sat there scratching my head - fact sheet? What is a fact sheet? And whatever it is, why can't we make one for ND?
So I wrote back. What is a fact sheet? What are the criteria which determine which diseases get to have a fact sheet? What can we, the Norrie community, do in order to have a fact sheet about ND? Etc.
This time the line went dead. No reply.
Silly me with my silly questions.
Do you know why there is no mention of Norrie disease on the NEI website? If so, I would love to hear from you. As always, I can be reached at maladiedenorrie@gmail.com.
Till next time then,
Meenu.
PS: The aforementioned tab on “A-Z Diseases and Disorders” appears to have been replaced by “Search A-Z” on the NEI website (just below the search box on the top right corner).
Wednesday, May 10, 2017
All the better to hear you with, my dear
The hearing loss associated with Norrie Disease (ND) is a particularly nasty symptom of this genetic disorder.
A blind person relies heavily on his/her sense of hearing for performing everyday tasks. Additionally, music is a favorite pastime for most blind people. To make matters worse, the hearing loss in ND does not present a stable situation, being progressive in nature.
Past research (Rehm et al., 2002) has established that in ND the damage to the ear happens in a part of the inner ear called the stria vascularis.
Now, I have heard of the cochlea, but what on earth is the stria vascularis?
A bit of of reading reveals that the stria vascularis is a highly vascular tissue within the cochlea.
Ok, that makes sense to me. The problem of ND is essentially a problem with the development and/or maintenance of the fine network of blood vessels which distribute blood within tissues (microvasculature). So ND would cause a loss of blood vessels in the stria vascularis, which would prevent the stria from doing whatever it is supposed to do, which would somehow result in a loss of hearing.
This is all a bit vague. I like digging deep into problems, so I read a bit more. I present a more in-depth explanation of the functioning of the stria vascularis in the boxed section below, which you may skip if you wish.
A blind person relies heavily on his/her sense of hearing for performing everyday tasks. Additionally, music is a favorite pastime for most blind people. To make matters worse, the hearing loss in ND does not present a stable situation, being progressive in nature.
Past research (Rehm et al., 2002) has established that in ND the damage to the ear happens in a part of the inner ear called the stria vascularis.
Now, I have heard of the cochlea, but what on earth is the stria vascularis?
A bit of of reading reveals that the stria vascularis is a highly vascular tissue within the cochlea.
Ok, that makes sense to me. The problem of ND is essentially a problem with the development and/or maintenance of the fine network of blood vessels which distribute blood within tissues (microvasculature). So ND would cause a loss of blood vessels in the stria vascularis, which would prevent the stria from doing whatever it is supposed to do, which would somehow result in a loss of hearing.
This is all a bit vague. I like digging deep into problems, so I read a bit more. I present a more in-depth explanation of the functioning of the stria vascularis in the boxed section below, which you may skip if you wish.
The inner ear consists of a complex series of tubes, running through the temporal bone of the skull. The bony tubes (sometimes called the bony labyrinth) are filled with a fluid called perilymph (shown in orange in the diagram below). Within this bony labyrinth is a second series of tubes, filled with a fluid called endolymph (shown in blue). So, this is a tube within a tube – each tube containing a different fluid.
The cochlea is part of the inner ear. It is a spiral structure (clearly visible in the above diagram) which resembles a snail (In fact, the word cochlea is derived from the Greek word kokhlias for a snail).
Found on the lateral wall of the cochlea, the stria vascularis is a highly vascular tissue which produces the endolymph. It is one of the most highly vascularized tissues found in the adult mammalian body (Zdebik et al., 2009). So what’s the significance of this? Well, the two fluids – perilymph and endolymph – have different ionic concentrations. Endolymph is rich in potassium, whereas perilymph is rich in sodium. This gives rise to a positive voltage (called the endocochlear potential or EP) of 80 - 100 mV seen in the endolymphatic space of the cochlea. If the stria was unable to produce endolymph, that would result in a loss of EP. Here is a nice little analogy to explain EP: If you hold a brick up in the air, it has available (potential) energy: if you drop it, that potential will be turned into actual energy when the brick hits the floor and makes a sound and a dent. Electrochemical potential is also a kind of available energy, waiting for an opportunity to do work. In the cochlea, electrochemical potential arises because there are different charges within fluid compartments of the inner ear, creating a flow of electric current as they try to equalize. The cochlea – or to be more precise, the specialized sensory cells (hair cells) within the cochlea – is/are responsible for converting sounds which enter the ear canal, from mechanical vibrations into electrical signals (this process is called transduction), which are then carried to the brain by the auditory nerve. It is endocochlear potential which is the driving force or ‘battery’ for the cochlear hair cells. So loss of function of Norrin results in loss of blood vessels in the stria vascularis, which means the stria vascularis cannot do its job of producing endolymph properly, which results in loss of endocochlear potential, which leads to loss of transduction, which signifies loss of hearing. Got it! |
So if scientists could inject a good copy of the gene Ndp in the inner ear, could that stop the hearing from deteriorating? Better still, could that reverse the hearing loss which has already taken place?
Probably. Maybe.
Until fairly recently, no one knew how to deliver a gene to the inner ear. Since there are more than 300 genetic defects which are known to prevent the hair cells in the human inner ear from working properly, a lot of research is going on, on how to restore hearing when it is lost due to a genetic defect. The latest news is, that scientists have managed to restore hearing in a mouse model of Usher syndrome. They have managed to do this “by using a modified, non-pathogenic adeno-associated virus (Anc80L65), which is introduced into the ear by way of a "Trojan Horse" to deliver genes to restore the functionality of the damaged hair cells” [Source: ScienceDaily
But there is still a long way to go. Translating this research from mouse to humans is going to take some time. The inner ear in humans is located within a cavity in the temporal bone of the skull. The temporal bone is one of the densest bones in the human body. How to deliver the virus to the human cochlea is one of the questions which remains to be answered.
And then the research needs to be applied to the specific case of Norrie disease. To the best of my knowledge, thus far there is no research specifically targeting the hearing loss associated with ND.
However, I do know that the Norrie Disease Association is trying very hard to get some research started on this symptom of Norrie disease, so I am keeping my fingers crossed.
Fingers crossed, toes crossed, everything crossed.
Till next time then,
Meenu.
Probably. Maybe.
Until fairly recently, no one knew how to deliver a gene to the inner ear. Since there are more than 300 genetic defects which are known to prevent the hair cells in the human inner ear from working properly, a lot of research is going on, on how to restore hearing when it is lost due to a genetic defect. The latest news is, that scientists have managed to restore hearing in a mouse model of Usher syndrome. They have managed to do this “by using a modified, non-pathogenic adeno-associated virus (Anc80L65), which is introduced into the ear by way of a "Trojan Horse" to deliver genes to restore the functionality of the damaged hair cells” [Source: ScienceDaily
But there is still a long way to go. Translating this research from mouse to humans is going to take some time. The inner ear in humans is located within a cavity in the temporal bone of the skull. The temporal bone is one of the densest bones in the human body. How to deliver the virus to the human cochlea is one of the questions which remains to be answered.
And then the research needs to be applied to the specific case of Norrie disease. To the best of my knowledge, thus far there is no research specifically targeting the hearing loss associated with ND.
However, I do know that the Norrie Disease Association is trying very hard to get some research started on this symptom of Norrie disease, so I am keeping my fingers crossed.
Fingers crossed, toes crossed, everything crossed.
Till next time then,
Meenu.
Monday, March 27, 2017
The genetic lottery
Dedication: For dearest Ruzbeh, and for each and every family out there that has ever felt despair and bewilderment at their (Norrie) child’s neurological/cognitive problems.
Acknowledgements: I thank the following scientists:
- Prof. Yvonne Jones and Dr. Tao-Hsin Chang, Division of Structural Biology, Wellcome Trust Centre for Human Genetics, University of Oxford, for taking the time and trouble to answer all my questions, and for permission to use figure 8B from Chang et al., 2015
- Prof. Jeremy Nathans, Professor of Molecular Biology and Genetics, of Neuroscience and of Ophthalmology, Johns Hopkins University School of Medicine, for providing clarifications to my queries related to Smallwood et al., 2007
- Dr. Jiyuan Ke, Research Scientist, Van Andel Research Institute, for permission to use figures 2A, 2B, and 2C from Ke et al., 2013
I would also like to thank families who have provided me with data pertaining to their (Norrie) child.
Here’s the thing – in Norrie Disease (ND) related literature, the number of cases presenting brain related problems (cognitive impairment, autistic traits, psychosis) is listed as up to 30-50%. This is a not a small percentage. Yet every once in a while, I get asked - How do you know your son’s mental problems are related to Norrie Disease? It might be something else.
An image of Ruzbeh – this amazingly intelligent person who, equally amazingly, is …….what? …….cognitively impaired? ...........intellectually impaired? …......has sensory integration problems? ………..I don’t even know what name I should give to it! – appears in my mind and I find myself saying - I don’t know that they are (related), I just assumed that they are.
Well, my assumption is hardly scientific proof, is it?
And so it was that I decided to take out some time to study Ruzbeh’s genetic mutation. Ruzbeh’s mutation is a missense mutation, which is the simplest type of mutation possible. A missense mutation implies that one amino acid (just one) of the protein has got substituted by another amino acid.
I requested members of the online Norrie support group for additional missense ND mutations, so that I could analyze all the mutations together for comparison.
I read through three research articles which, directly or indirectly, deal with missense mutations of Norrin. These are: Smallwood et al, 2007, Ke et al., 2013, and Chang et al., 2015.
I was amazed at what I learnt.
Before I get to my findings though, I thought you might like to read the following:
- a recap of some basic facts about genes, proteins, and amino acids
- definition of a missense mutation
- the composition, structure, and functioning of the protein Norrin
An organism's genetic blueprint is encoded by the molecule called Deoxyribonucleic acid (DNA). DNA is a linear molecule composed of four types of smaller chemical molecules called nucleotide bases: adenine (A), cytosine (C), guanine (G), and thymine (T). The order of these bases is called the DNA sequence.
Segments of DNA that carry genetic information are called genes, and they are inherited by offspring from their parents during reproduction. Each gene is a coded description for making a particular protein. The sequence of bases in the gene determines the protein that it codes for.

Proteins perform a large chunk of the important jobs a cell needs to do. They make up our muscles, hair, nails; they make chemical reactions happen; they digest our food. Protein molecules are long chains of amino acids that are folded into a three-dimensional shape. The shape a protein takes is incredibly important for its function. Think about it - if a key has the wrong shape, it can't open a lock, right? The same is true for a protein and its function.
Amino acids are small molecules that are the building blocks of proteins. There are 20 amino acids & they have names like leucine, methionine, tryptophan, etc. Each amino acid is denoted either by a 3 letter abbreviation or by a 1 letter code. So leucine is Leu or L, methionine is Met or M, and tryptophan is Trp or W.
The genetic code is a set of three-letter combinations (called codons) of the four nucleotide bases (A, C, T, G), each of which corresponds to a specific amino acid or stop signal.
There are 64 possible permutations, or combinations, of three-letter sequences that can be made from the four nucleotides. Of these 64 codons, 61 represent amino acids, and three are stop signals. Stop codons serve as a signal that the end of the chain has been reached during protein synthesis.
Given below is a table depicting the genetic code.

The colors highlight the fact that most amino acids have more than one code. For example, Serine (Ser) has 6 codes.
The 1 letter code corresponding to each amino acid is as shown below:
| Alanine | Ala | A |
| Arginine | Arg | R |
| Asparagine | Asn | N |
| Aspartic Acid | Asp | D |
| Cysteine | Cys | C |
| Glutamine | Gln | Q |
| Glutamic Acid | Glu | E |
| Glycine | Gly | G |
| Histidine | His | H |
| Isoleucine | Ile | I |
| Leucine | Leu | L |
| Lysine | Lys | K |
| Methionine | Met | M |
| Phenylalanine | Phe | F |
| Proline | Pro | P |
| Serine | Ser | S |
| Threonine | Thr | T |
| Tryptophan | Trp | W |
| Tyrosine | Tyr | Y |
| Valine | Val | V |
When amino acids combine to form a protein, they are also referred to as amino acid residues, or simply residues.
As an example, consider the peptide (a peptide is simply a short chain of amino acids) Gly-Ser-Gln-Met-Val-Ile or GSQMVI. The 1st residue (G1) is the amino acid glycine (Gly), the 5th residue (V5) is the amino acid valine (Val), and so on.
Based on the propensity of an amino acid to be in contact with water, it may be classified as hydrophobic (low propensity to be in contact with water), neutral, or hydrophilic.
(2) Missense mutations [Source: Wikipedia]
A mutation is a change in a genetic sequence. Mutations include changes as small as the substitution of a single DNA building block, or nucleotide base, with another nucleotide base. Along with substitutions, mutations can also be caused by insertions, deletions, or duplications of DNA sequences. Mutations can be introduced due to mistakes made during DNA replication or due to exposure to mutagens, which are chemical and environmental agents that can introduce mutations in the DNA sequence.
A missense mutation is a genetic change which results in one amino acid of a protein getting substituted by another amino acid.
For example, a change in just one nucleotide base can result in a missense mutation, as shown below:

(3) A brief note on the composition, structure & function of Norrin
The Norrie Disease gene (Ndp) codes for the protein Norrin.
Norrin comprises of 133 residues. These are listed below:
KTDSSFIMDSDPRRCMRHHYVDSISHPLYKCSSKMVLLARCEGHCSQASRSEPLVSFSTVLKQPFRSSCHCCRPQTSKLKALRLRCSGGMRLTATYRYILSCHCEECNS
Norrin structure
The crystal structure of human Norrin reveals a dimer formation. A dimer consists of two structurally similar molecules, each of which is referred to as a monomer. Norrin is a homodimer, which means that the two monomers are identical. The following is a ribbon diagram of the Norrin dimer consisting of two identical monomers in green and magenta:

[Source: Ke et al., 2013 Figure 2B]
Each Norrin monomer contains 11 cysteines (Cys or C) – C39, C55, C65, C69, C93, C95, C96, C110, C126, C128 & C131. These are shown in red below:
KTDSSFIMDSDPRRCMRHHYVDSISHPLYKCSSKMVLLARCEGHCSQASRSEPLVSFSTVLKQPFRSSCHCCRPQTSKLKALRLRCSGGMRLTATYRYILSCHCEECNS
Cysteine has a sulfur atom (the chemical formula for cysteine being C3H7NO2S). Sulfur atoms on two separate cysteine residues can bond with each other, forming a disulfide bridge or a disulphide bond.

Schematic representation of disulphide bonds in a protein [Source: Wikipedia]
Of the eleven cysteines of the Norrin monomer, six cysteines form three pairs of intramolecular (i.e. contained within the Norrin monomer) disulphide bonds (C39–C96, C65–C126, C69–C128) which result in the protein folding into a cystine knot structure.

Diagram depicting the cystine knot motif of Norrin. A: Amino acid sequence of norrin. Highlighted C's indicate cysteine residues involved in disulfide bonds which lead to the cystine knot structure. B: Structure of Norrin. The roman numerals label the cysteine residues involved in cystine knot disulfide bonds (connecting lines).
[Source: K Drenser et al., 2007]
Norrin function depends critically on the three pairs of cysteines that form the highly conserved trio of disulfide bonds shared among all cystine knot proteins (Smallwood et al, 2007).
Another pair of cysteines (C55–C110) forms a fourth intramolecular disulphide bond.
The following diagram shows all four intramolecular disulfide bonds (C39–C96, C65–C126, C69–C128, and C55–C110) in the Norrin monomer:
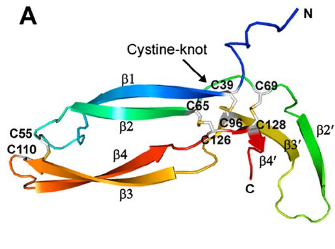
[Source: Ke et al., 2013 Figure 2A]
Additionally, there exist three intermolecular (i.e. between the two monomers of the Norrin dimer) disulphide bonds (C93–C95, C95–C93, and C131–C131). These disulphide bonds are important for the stability of the Norrin dimer.
The following is a diagram of the Norrin dimer, showing the three intermolecular disulfide bonds (C93–C95, C95–C93, and C131–C131):

[Source: Ke et al., 2013 Figure 2C]
In addtition to these three cysteine residues, the Norrin dimer interface comprises of numerous other residues. The dimeric interface of Norrin is important for its function.
Functioning of Norrin
The cells within an animal body need to be able to communicate with each other to coordinate many complex processes in the body, such as the formation of tissues and organs. One way in which the cells can communicate is through a pathway called Wnt signaling.
Norrin activates the Wnt signaling pathway by binding (attaching) to two other proteins/molecules - Frizzled-4 (also written as Fzd-4 or Fzd4) and Lrp5/6. For the purpose of this blog, we don’t really need to know a great deal about these molecules. Suffice it to say that Fzd4 and Lrp5/6 are proteins coded for by the genes FZD4, LRP5, and LRP6, in much the same way that Norrin is a protein coded for by the gene NDP. In scientific parlance, Fzd4 and Lrp5/6 are referred to as receptors of Norrin.
Norrin has distinct binding sites for Fzd4 and for Lrp5/6. The binding site for Fzd4 and a potential binding site for Lrp5/6 have been mapped out in Chang et al., 2015, as shown in the diagram below. The two concave shapes in the diagram represent the two monomers of Norrin.

Diagram depicting the various binding sites on Norrin. The two concave shapes represent the two monomers of Norrin. GAG (glycosaminoglycan) is yet another type of molecule which binds to the Norrin-Fzd4 complex. [Source: Chang et al., 2015]
The following table categorizes residues of Norrin based on the structure and/or function of the protein:
| Category | Residues |
| Cystine knot motif | C39, C65, C69, C96, C126, C128 |
| Fzd-4 binding site | D33, S44, P36, R38, residues 40-43, V45, D46, S47, M59, V60, L61, T100, K102, K104, A105, L106, Y120, Y122, L124, S125 |
| Lrp5/6 binding site | K54, R90, R97, G112, R121 |
| GAG binding site | K58, R107, R109, R115 |
| Dimer interface | Y44, I48, S49, H50, P51, residues 62-81, residues 83-85, P88, F89, S91, residues 93-96, P98, S101, L103, L108, residues 116-119, I123, C131, N132 |
| Other | Any residue of Norrin which is not listed in any of the above five categories |
(4) Findings
I had four mutations to analyze:
- C69Y (the 69th residue has got substituted by tyrosine in place of cysteine)
- R90C (the 90th residue has got substituted by cysteine in place of arginine)
- F81L (the 81st residue has got substituted by leucine in place of phenylalanine)
- G112Q (the 112th residue has got substituted by glutamine in place of glycine)
C69Y: This is Ruzbeh’s mutation. C69!! This is one of the six cysteines which form the disulphide bonds leading to the cystine knot motif of Norrin. I presume this mutation would dramatically alter the Norrin signaling activity.
R90C, F81L, G112Q: These mutations were provided by families from the Norrie support group. These are easily identified as mutations on the (potential) Lrp5/6 binding site (residues R90 and G112) and on the dimer interface (residue F81).
| Missense mutation | Category of residue* | Age of person / Symptoms seen** |
| C69Y | One of the six cysteines which form the cystine knot motif of Norrin. | 18 years (Ruzbeh).
Congenital blindness, hearing loss, autistic behavior, cognitive impairment, few psychotic episodes, two seizures. |
| R90C | Lrp5/6 binding site | 8 years.
Congenital blindness, few language difficulties, sensory sensitive. |
| F81L | Dimer interface | 4 years.
Congenital blindness, developmental delay, mild learning difficulties. |
| G112Q | Lrp5/6 binding site | 22 years.
Blind in one eye due to retina detachment at age 2 ½ years old. 20/30 vision in other eye post vitrectomy surgery at age 4. |
* Source: Chang et al., 2015
** Data provided by families
So far so good.
I wanted to go a step further and find some quantitative data for these mutations. In separate communications from Prof. Nathans and from Prof. Jones, I have come to know that quantitative data for Norrin’s signaling activity for different mutations, does not exist. However, they did have this to say:
C69Y (Ruzbeh’s mutation):
Prof. Nathans agrees that C69 being a critical residue of Norrin, this mutation would result in a severe loss of function of the protein Norrin.
Prof. Jones: Not all residues in a protein have equally important roles. Some missense mutations are likely to have more effect because of the position of the residue in the 3D structure of the protein and/or because of the extent of the change the missense mutation makes to the properties of the residue. For example, a particular residue can be essential for a protein to fold into its correct 3D shape, and/or to maintain that shape. This category of residue is important because if the protein does not have the correct overall shape it cannot work properly. The six cysteine residues which form the cystine-knot motif of Norrin do so because, uniquely, one cysteine can bond to another cysteine to form a disulphide bond, and the three disulphide bonds of the cystine-knot motif are what locks one copy of Norrin into its correct 3D shape.
Sadly, the extent of your son’s symptoms is consistent with this being a missense mutation affecting the cystine-knot motif and so stopping the protein forming the correct 3D shape and therefore stopping its ability to bind to both its receptors.
R90C, F81L, G112Q
Prof. Jones: Norrin works as a dimer so the residues that contribute to the dimer interface between the two copies of Norrin are important.
Three cysteine residues per copy of Norrin hold two copies together by forming disulphide bonds at the dimer interface. Because of the unique ability of cysteine to form a disulphide bond, and because of the role of disulphide bridges in stabilising the correct 3D structure of Norrin, missense mutations of these residues are likely to be serious.
How serious missense mutations are of other residues at the dimer interface, or of the residues that Norrin uses to bind to its receptors Fzd4 and Lrp5/6, depends on whether the missense mutation makes a big change to essential properties of the residue.
R90C and G112Q both result in big changes to the properties of the affected residues but we do not know how these particular properties contribute to the binding of Norrin to Lrp5/6 because we do not have the structure of Norrin bound to Lrp5/6. Also, without a structure, we do not know whether the residue is at the centre of the binding site, often a residue at the centre of the binding site will make more of a contribution than one at the edge. The only thing we can say now is that we do not expect these residues to affect the overall 3D shape of the protein or its ability to bind to Lrp5/6.
If we take a residue that contributes to the ability of Norrin to bind to one of its receptors, or to
form the dimer interface, because it is a ‘hydrophobic’ residue (i.e. ‘sticky’) then a missense mutation that changes the residue to one that is slightly different in size, but still hydrophobic/sticky, might not have such a serious effect – the dimer may be slightly distorted or the binding to the receptor may be weaker, so the signaling activity is reduced, but it is not lost completely.
F81L is at the dimer interface of Norrin. F (phenylalanine) is a sticky/hydrophobic residue, and so is L (leucine), albeit F is a bit bigger than L. We think that the dimer is likely to still form but it may be a little distorted so the binding of F81L Norrin to its receptors may be affected because it is not quite the same 3D shape as usual.
So there you have it. In black and white.
And I finally know. The reason why.
Till next time then,
Meenu.
Wednesday, February 15, 2017
More answers, more questions
Today’s post is about my struggle to find answers to questions such as the following:
I decided to get in touch with Prof. Jeremy Nathans, professor of molecular biology and genetics, of neuroscience, and of ophthalmology at the Johns Hopkins University School of Medicine, in order to try and make sense of all that I have read so far in relation to the above.
Before I present to you the ensuing conversation, here is a background to the questions posed by me:
From the research that I have read, it seems that
Further, traditionally the function of the cerebellum has been thought of as being motor control, but this view is being increasingly challenged. I cite a few examples:
And so, on to the ‘conversation’ (which took place via email) with Prof. Nathans, presented here with his kind permission.
Qs: From the research done by you and your colleagues, would it be correct to infer that a leaky BBB is present in all cases of Norrie Disease (ND), and that the difference amongst various cases is primarily that of the extent of leakiness?
Ans: I do not think that there is any direct data on BBB integrity in humans with ND. Also, the BBB in most of the brain is protected from loss of Norrin by a second redundant signaling system that uses Wnt7a and Wnt7b (at least in mice). The retina does not have the back-up system (in mice).
Qs: While there is no direct data on BBB integrity on humans, I thought one could make inferences based on lab based research on animals?
Also, when you say "most of the brain", does this exclude the cerebellum & olfactory bulb?
Ans: Extrapolating from mice to humans is usually OK, but not always. Yes, the cerebellum and olfactory bulb show a mild BBB defect when Norrin is absent - so for these two locations the BBB is not completed covered by the Wnt7a and Wnt7b system.
Qs: If one wanted to know if a similar BBB defect exists in the case of ND in humans, what would be the way to determine this? Would it show up in an MRI for example?
Ans: That is a very good question, and I do not know the answer. I think we would have to ask the neuro-radiologists.
Qs: I rephrase my very first question. From the research done by you and your colleagues, would it be correct to infer that a leaky BBB is present in the cerebellum and the olfactory bulb in all cases of a loss of function mutation in the Norrie gene (Ndp) in mice?
Ans: Phrased that way, the answer is yes. It is very consistent in mice.
Qs: Was there an opportunity to observe whether the extent of leakiness varied across different mutations of Ndp?
Ans: No, we only have one mouse mutation and it completely eliminates Norrin.
Qs: One of the symptoms of ND is neurological disorders. In ND related literature this is reported in up to 30-50% of cases. IF – and I realize this is a big if - the same BBB defects that are seen in Norrie knockout mice be found in patients of ND as well, then this could help explain these symptoms. Since not all cases of ND present these symptoms, I think it would be interesting to see if the extent of BBB leakiness varies across different Ndp mutations. Would you care to weigh in on this?
Ans: That is a very plausible idea.
Qs: There has been mention in ND related literature of epilepsy & seizures, and we see this in several cases of the present day online Norrie community also.
Studies have shown that BBB dysfunction in the hippocampus is one of the many causes of epilepsy (van EA Vliet et al., 2014).
While I could not find any mention of the hippocampus in your study, which used mice, I did find another, older study (Hartzer et al., 1999) which tells us that the gene Ndp is expressed in the hippocampus (& also the cortex!) of the rabbit brain (………… In order to further determine the role of the Norrie disease gene, we studied the distribution pattern of its mRNA in the retina and in brain by in situ hybridization. …….. High expression levels were also observed in the cerebellar granular layer, hippocampus, olfactory bulb, cortex, and epithelium of the rabbit brain. …….).
So the logical conclusion would be that we cannot rule out the possibility that the gene Ndp may be expressed in the human hippocampus as well as the cortex? Would you agree?
Ans: The mouse data shows that Ndp is likely expressed widely (nearly everywhere) in the brain.
Qs: If Ndp is expressed widely in the brain, but it plays a role in BBB maintenance primarily in the cerebellum and olfactory bulb, then do we know what role it plays in the other parts of the brain?
Ans: It appears that signaling by a second system (based on proteins called Wnt7a and Wnt7b) functions in parallel to Norrin signaling in many parts of the brain (but not the retina) and in those regions it covers up the deficiencies that would be seen if Norrin alone were responsible. This is all based on work in mice.
Qs: A later study (Luhmann et al., 2008) speaks of reduced vessel density in the cerebellum of Ndph knockout mice. Is this consistent with your findings?
Ans: We agree. There is a subtle reduction in vessel density in cerebellum in Ndp KO mice.
Qs: Is the problem of how to restore the Norrin signaling in order to restore the BBB for ND patients under active consideration by your lab? If yes, then would this involve delivering the protein Norrin to the brain?
Ans: We have not pursued that line of investigation. Delivering therapeutic proteins to the brain is challenging because the BBB is a barrier to entry. Many biotech and pharmaceutical companies are working on this.
Qs: In speaking to another researcher, I had come to know that in the past it has been difficult to obtain a consistently good quality of Norrin - the quality varies over batches - something to do with the cystine knot structure of the protein. Would you know if this problem has been resolved or is this yet another hurdle to be overcome?
Ans: I have not tried using commercial Norrin protein, but in our own lab’s experience it is difficult to produce in large quantities. However, at least one research group appears to have solved this problem: Yvonne Jones at Oxford University has made large amounts of pure Norrin for structural studies.
I thank Prof. Nathans for his time and patience.
So where does this all take me?
I think the neurological issues seen in some cases of ND are related to the defects seen in the cerebellum of Norrie knockout mice.
I think the reason why some cases of ND present neurological issues while others do not, has something to do with the particular mutation – that is to say that it has to do with the extent of damage caused by a particular mutation.
I think that while there is no BBB defect seen in the hippocampus of Norrie knockout mice, the jury is still out on whether the epilepsy & seizures seen in some cases of ND are related to ND or not, because when I look at individual cases in our present day online Norrie community, in almost all - if not all – cases, the person who has experienced seizures, also presents neurological issues. I do not remember an instance where this is not the case. (The converse is not true – not everyone with neurological issues has experienced epilepsy, but this could once again be about the extent of damage caused by a particular mutation).
There is more work to be done here.
Till next time then,
Meenu.
- Are the neurological issues seen in some cases of ND linked to the Blood Brain Barrier (BBB) leakiness of the cerebellum found in ND related research?
- If yes, then why is it that some cases of ND have neurological issues while others do not?
- Are the epilepsy & seizures reported in some cases of ND an outcome of ND?
I decided to get in touch with Prof. Jeremy Nathans, professor of molecular biology and genetics, of neuroscience, and of ophthalmology at the Johns Hopkins University School of Medicine, in order to try and make sense of all that I have read so far in relation to the above.
Before I present to you the ensuing conversation, here is a background to the questions posed by me:
From the research that I have read, it seems that
- there is a Blood Brain Barrier (BBB) dysfunction in the cerebellum and the olfactory bulb in ND (Nathans et al., 2014)
- there is a reduction in blood vessel density in the cerebellum in ND (Luhmann et al., 2008)
- the gene Ndp is expressed in the cerebellum, hippocampus, olfactory bulb, cortex, and epithelium of the brain (Hartzer et al., 1999)
- BBB dysfunction in the hippocampus is one of the many causes of epilepsy (van EA Vliet et al., 2014)
Further, traditionally the function of the cerebellum has been thought of as being motor control, but this view is being increasingly challenged. I cite a few examples:
- Cerebellar function was once believed to be motor-specific, but newer findings suggest the cerebellum is also involved in higher-level brain processing. Functional imaging studies have also shown cerebellar activation in relation to language, attention, and mental imagery. [Source: Boundless, 11 Oct. 2016. “Functions of the Cerebellum” ]
- The traditional teaching that the cerebellum is purely a motor control device no longer appears valid, if, indeed, ever it was. There is increasing recognition that the cerebellum contributes to cognitive processing and emotional control in addition to its role in motor coordination.[Source: Brain, Feb. 2006. “Cognition, emotion and the cerebellum”]
And so, on to the ‘conversation’ (which took place via email) with Prof. Nathans, presented here with his kind permission.
Qs: From the research done by you and your colleagues, would it be correct to infer that a leaky BBB is present in all cases of Norrie Disease (ND), and that the difference amongst various cases is primarily that of the extent of leakiness?
Ans: I do not think that there is any direct data on BBB integrity in humans with ND. Also, the BBB in most of the brain is protected from loss of Norrin by a second redundant signaling system that uses Wnt7a and Wnt7b (at least in mice). The retina does not have the back-up system (in mice).
Qs: While there is no direct data on BBB integrity on humans, I thought one could make inferences based on lab based research on animals?
Also, when you say "most of the brain", does this exclude the cerebellum & olfactory bulb?
Ans: Extrapolating from mice to humans is usually OK, but not always. Yes, the cerebellum and olfactory bulb show a mild BBB defect when Norrin is absent - so for these two locations the BBB is not completed covered by the Wnt7a and Wnt7b system.
Qs: If one wanted to know if a similar BBB defect exists in the case of ND in humans, what would be the way to determine this? Would it show up in an MRI for example?
Ans: That is a very good question, and I do not know the answer. I think we would have to ask the neuro-radiologists.
Qs: I rephrase my very first question. From the research done by you and your colleagues, would it be correct to infer that a leaky BBB is present in the cerebellum and the olfactory bulb in all cases of a loss of function mutation in the Norrie gene (Ndp) in mice?
Ans: Phrased that way, the answer is yes. It is very consistent in mice.
Qs: Was there an opportunity to observe whether the extent of leakiness varied across different mutations of Ndp?
Ans: No, we only have one mouse mutation and it completely eliminates Norrin.
Qs: One of the symptoms of ND is neurological disorders. In ND related literature this is reported in up to 30-50% of cases. IF – and I realize this is a big if - the same BBB defects that are seen in Norrie knockout mice be found in patients of ND as well, then this could help explain these symptoms. Since not all cases of ND present these symptoms, I think it would be interesting to see if the extent of BBB leakiness varies across different Ndp mutations. Would you care to weigh in on this?
Ans: That is a very plausible idea.
Qs: There has been mention in ND related literature of epilepsy & seizures, and we see this in several cases of the present day online Norrie community also.
Studies have shown that BBB dysfunction in the hippocampus is one of the many causes of epilepsy (van EA Vliet et al., 2014).
While I could not find any mention of the hippocampus in your study, which used mice, I did find another, older study (Hartzer et al., 1999) which tells us that the gene Ndp is expressed in the hippocampus (& also the cortex!) of the rabbit brain (………… In order to further determine the role of the Norrie disease gene, we studied the distribution pattern of its mRNA in the retina and in brain by in situ hybridization. …….. High expression levels were also observed in the cerebellar granular layer, hippocampus, olfactory bulb, cortex, and epithelium of the rabbit brain. …….).
So the logical conclusion would be that we cannot rule out the possibility that the gene Ndp may be expressed in the human hippocampus as well as the cortex? Would you agree?
Ans: The mouse data shows that Ndp is likely expressed widely (nearly everywhere) in the brain.
Qs: If Ndp is expressed widely in the brain, but it plays a role in BBB maintenance primarily in the cerebellum and olfactory bulb, then do we know what role it plays in the other parts of the brain?
Ans: It appears that signaling by a second system (based on proteins called Wnt7a and Wnt7b) functions in parallel to Norrin signaling in many parts of the brain (but not the retina) and in those regions it covers up the deficiencies that would be seen if Norrin alone were responsible. This is all based on work in mice.
Qs: A later study (Luhmann et al., 2008) speaks of reduced vessel density in the cerebellum of Ndph knockout mice. Is this consistent with your findings?
Ans: We agree. There is a subtle reduction in vessel density in cerebellum in Ndp KO mice.
Qs: Is the problem of how to restore the Norrin signaling in order to restore the BBB for ND patients under active consideration by your lab? If yes, then would this involve delivering the protein Norrin to the brain?
Ans: We have not pursued that line of investigation. Delivering therapeutic proteins to the brain is challenging because the BBB is a barrier to entry. Many biotech and pharmaceutical companies are working on this.
Qs: In speaking to another researcher, I had come to know that in the past it has been difficult to obtain a consistently good quality of Norrin - the quality varies over batches - something to do with the cystine knot structure of the protein. Would you know if this problem has been resolved or is this yet another hurdle to be overcome?
Ans: I have not tried using commercial Norrin protein, but in our own lab’s experience it is difficult to produce in large quantities. However, at least one research group appears to have solved this problem: Yvonne Jones at Oxford University has made large amounts of pure Norrin for structural studies.
I thank Prof. Nathans for his time and patience.
So where does this all take me?
I think the neurological issues seen in some cases of ND are related to the defects seen in the cerebellum of Norrie knockout mice.
I think the reason why some cases of ND present neurological issues while others do not, has something to do with the particular mutation – that is to say that it has to do with the extent of damage caused by a particular mutation.
I think that while there is no BBB defect seen in the hippocampus of Norrie knockout mice, the jury is still out on whether the epilepsy & seizures seen in some cases of ND are related to ND or not, because when I look at individual cases in our present day online Norrie community, in almost all - if not all – cases, the person who has experienced seizures, also presents neurological issues. I do not remember an instance where this is not the case. (The converse is not true – not everyone with neurological issues has experienced epilepsy, but this could once again be about the extent of damage caused by a particular mutation).
There is more work to be done here.
Till next time then,
Meenu.
Wednesday, January 18, 2017
Norrie Disease and Epilepsy – Is there a link?
In my very first blog post, I had put forward a hypothesis that the epilepsy seen in some cases of Norrie Disease (ND) could be linked to the Blood Brain Barrier (BBB) dysfunction which occurs in ND. I had also mentioned a research article (van EA Vliet et al., 2014) which links BBB dysfunction with epilepsy.
Since epilepsy happens to a great many people, not just to patients of ND, I set out in search of an answer to the question - is there really a link between ND and epilepsy?
I thought I would start by educating myself a bit more on the link between the BBB and epilepsy, by reaching out to Dr. Vliet, one of the authors of the above mentioned study. Dr. Vliet is a member of the Faculty of Science, University of Amsterdam.
Dr. Vliet has very kindly answered my questions with respect to epilepsy and the BBB, and has also been kind enough to allow me to share my learnings with the readers of this blog.
He would, however, like readers to note that he is not a clinician, but a basic scientist, that he is not an expert on Norrie Disease, and that his answers are from the perspective of preclinical work done by him.
Qs1: Would a leaky blood brain barrier always lead to epilepsy? If not, then do we know which aspect of BBB dysfunction gives rise to epilepsy?
The reason why I ask is because only a small percentage (perhaps 5-10%) of ND cases have seizures. On the other hand, 30-50% of cases are reported to have mental problems ranging from mild to severe (cognitive impairment, autistic behavior, psychosis), and it is my assumption (which could of course be wrong) that these problems are caused by BBB dysfunction.
Ans.: A leaky BBB does not always lead to epilepsy. Most likely the amount/extent of leakage, the brain region in which in occurs, and other pathological factors that may occur (e.g. gliosis, neuronal loss etc.) are also important. From preclinical epilepsy models we see that the larger the extent of leakage, particularly in the hippocampus/parahippocampus (temporal lobe), the more likely the chance to develop epilepsy. Lastly, BBB leakage does not immediately lead to seizures, it takes time (in animals a few weeks).
Qs2: Conversely, is epilepsy always caused by BBB dysfunction? Apparently epilepsy is the 4th most common neurological disorder, so I wonder if it is wrong on my part to try and link the BBB dysfunction of ND to the epilepsy seen in ND.
Ans.: There are many factors that can lead to epilepsy (e.g. tumors, inflammation, infection, injury etc.), BBB leakage is one of them. So epilepsy is not always caused by BBB leakage.
Qs3: If we were to conclude that a person’s epilepsy was caused by BBB dysfunction, then would restoring the BBB integrity cure the problem of epilepsy?
Ans.: That's a very good point. Therefore, we aim to restore BBB function and thereby hope to inhibit or prevent epilepsy. In preclinical models this seems to be the case but it has not been tested in people with epilepsy.
I thank Dr. Vliet for his valuable time.
Dear reader, I must now apply what I have learnt, to the case of Norrie disease.
Past research (Hartzer et al., 1999) has established that the Norrie disease gene (NDP) is expressed in the hippocampus: …… In order to further determine the role of the Norrie disease gene, we studied the distribution pattern of its mRNA in the retina and in brain by in situ hybridization. …….. High expression levels were also observed in the cerebellar granular layer, hippocampus, olfactory bulb, cortex, and epithelium of the rabbit brain. …….
However, later research done by Prof. Jeremy Nathans and colleagues (Nathans et al., 2014) suggests that the protein (Norrin) which is encoded by the gene does not play a role in BBB maintenance in the hippocampus: …… mutation of Ndp produces only a relatively mild loss of BBB integrity that is confined to the cerebellum and olfactory bulb. ……
This study does talk of BBB dysfunction in other parts of the brain in the case of a mutation of the NDP gene, but that is under very special circumstances (other genes being mutated in combination with the NDP gene), but even then there is no mention of the hippocampus.
So if the NDP gene is expressed in the hippocampus, then it must be doing something there. What is that something? Also, we know nothing about the extent of damage in the brain caused by various mutations of the Norrie gene. What if the BBB dysfunction in those cases of ND where epilepsy has happened is more severe than in other cases? Would that have a bearing on the question in focus?
There is more work to be done here.
Till next time then,
Meenu.
Since epilepsy happens to a great many people, not just to patients of ND, I set out in search of an answer to the question - is there really a link between ND and epilepsy?
I thought I would start by educating myself a bit more on the link between the BBB and epilepsy, by reaching out to Dr. Vliet, one of the authors of the above mentioned study. Dr. Vliet is a member of the Faculty of Science, University of Amsterdam.
Dr. Vliet has very kindly answered my questions with respect to epilepsy and the BBB, and has also been kind enough to allow me to share my learnings with the readers of this blog.
He would, however, like readers to note that he is not a clinician, but a basic scientist, that he is not an expert on Norrie Disease, and that his answers are from the perspective of preclinical work done by him.
Qs1: Would a leaky blood brain barrier always lead to epilepsy? If not, then do we know which aspect of BBB dysfunction gives rise to epilepsy?
The reason why I ask is because only a small percentage (perhaps 5-10%) of ND cases have seizures. On the other hand, 30-50% of cases are reported to have mental problems ranging from mild to severe (cognitive impairment, autistic behavior, psychosis), and it is my assumption (which could of course be wrong) that these problems are caused by BBB dysfunction.
Ans.: A leaky BBB does not always lead to epilepsy. Most likely the amount/extent of leakage, the brain region in which in occurs, and other pathological factors that may occur (e.g. gliosis, neuronal loss etc.) are also important. From preclinical epilepsy models we see that the larger the extent of leakage, particularly in the hippocampus/parahippocampus (temporal lobe), the more likely the chance to develop epilepsy. Lastly, BBB leakage does not immediately lead to seizures, it takes time (in animals a few weeks).
Qs2: Conversely, is epilepsy always caused by BBB dysfunction? Apparently epilepsy is the 4th most common neurological disorder, so I wonder if it is wrong on my part to try and link the BBB dysfunction of ND to the epilepsy seen in ND.
Ans.: There are many factors that can lead to epilepsy (e.g. tumors, inflammation, infection, injury etc.), BBB leakage is one of them. So epilepsy is not always caused by BBB leakage.
Qs3: If we were to conclude that a person’s epilepsy was caused by BBB dysfunction, then would restoring the BBB integrity cure the problem of epilepsy?
Ans.: That's a very good point. Therefore, we aim to restore BBB function and thereby hope to inhibit or prevent epilepsy. In preclinical models this seems to be the case but it has not been tested in people with epilepsy.
I thank Dr. Vliet for his valuable time.
Dear reader, I must now apply what I have learnt, to the case of Norrie disease.
Past research (Hartzer et al., 1999) has established that the Norrie disease gene (NDP) is expressed in the hippocampus: …… In order to further determine the role of the Norrie disease gene, we studied the distribution pattern of its mRNA in the retina and in brain by in situ hybridization. …….. High expression levels were also observed in the cerebellar granular layer, hippocampus, olfactory bulb, cortex, and epithelium of the rabbit brain. …….
However, later research done by Prof. Jeremy Nathans and colleagues (Nathans et al., 2014) suggests that the protein (Norrin) which is encoded by the gene does not play a role in BBB maintenance in the hippocampus: …… mutation of Ndp produces only a relatively mild loss of BBB integrity that is confined to the cerebellum and olfactory bulb. ……
This study does talk of BBB dysfunction in other parts of the brain in the case of a mutation of the NDP gene, but that is under very special circumstances (other genes being mutated in combination with the NDP gene), but even then there is no mention of the hippocampus.
So if the NDP gene is expressed in the hippocampus, then it must be doing something there. What is that something? Also, we know nothing about the extent of damage in the brain caused by various mutations of the Norrie gene. What if the BBB dysfunction in those cases of ND where epilepsy has happened is more severe than in other cases? Would that have a bearing on the question in focus?
There is more work to be done here.
Till next time then,
Meenu.
Wednesday, December 21, 2016
Moving on
This post almost got postponed to the New Year, given that this week has been so, so tiring in the wake of Ruzbeh’s epileptic attack. But I couldn’t bear the thought of letting Norrie disease get the better of me, so here goes…
In the aftermath of my discussion with the retina specialist (read my previous blogpost), I felt a pall of gloom descend upon me. What now? I wondered.
For a while I pondered reading up on other symptoms of Norrie disease (ND), something that I’ve been putting off for sheer lack of time.
And yet. And yet.
My thoughts were in turmoil: What about all the hours I spent reading stuff. What about my conviction that one day it will be possible to reverse the blindness associated with ND.
Research is not about saying it can’t be done, research is about saying it needs to be done, and then finding a way to do it. Or so I believe.
And then my thoughts turned to the NEI (National Eye Institute). If you have never heard of the NEI, this is what it says on their website:
As part of the federal government’s National Institutes of Health (NIH), the National Eye Institute’s mission is to “conduct and support research, training, health information dissemination, and other programs with respect to blinding eye diseases, visual disorders, mechanisms of visual function, preservation of sight, and the special health problems and requirements of the blind.” The NEI was established in 1968. It has a budget of approximately $675 million (FY2014).
What if they could gather a few researchers for a small problem solving session? The objectives of this meeting would be the following:
i) To lay down step by step, what needs to happen for vision to be restored in a case of ND
ii) To identify from step (i), those areas where research is currently going on & those which are not the subject of current research
iii) To identify from step (ii), those areas where some ND specific research needs to be done in conjunction with ongoing research
If funds need to be arranged for this brainstorming to happen, then I would be happy to try and secure them. I don’t know how, but I’ll cross that bridge when I come to it.
My point is this: It is true that I can’t put a million dollars on the table for ND related research, but if we could define the way to solve the problem of vision (or lack of), then maybe I could find a way to obtain funds for individual projects.
So I went ahead and hammered out a letter to this effect.
Then I pondered for a bit about who to address it to. I figured I would send it to Dr. Paul Sieving, Director, NEI.
Then I pondered a bit more – what if the Norrie Disease Association (NDA) has had some dealings with the NEI in the past? In that case maybe they could tell me who to get in touch with. So I wrote to John Miller, President, NDA. No, the NDA has not been in touch with the NEI.
And yet I hesitated. I really want this brainstorming to happen. No more no’s. Perhaps I ought to route my request through the NDA? Perhaps that would strengthen my case? Hmmmm. Yes, I think I will try the NDA route. Let’s see where it leads.
Till next time then,
Meenu.
In the aftermath of my discussion with the retina specialist (read my previous blogpost), I felt a pall of gloom descend upon me. What now? I wondered.
For a while I pondered reading up on other symptoms of Norrie disease (ND), something that I’ve been putting off for sheer lack of time.
And yet. And yet.
My thoughts were in turmoil: What about all the hours I spent reading stuff. What about my conviction that one day it will be possible to reverse the blindness associated with ND.
Research is not about saying it can’t be done, research is about saying it needs to be done, and then finding a way to do it. Or so I believe.
And then my thoughts turned to the NEI (National Eye Institute). If you have never heard of the NEI, this is what it says on their website:
As part of the federal government’s National Institutes of Health (NIH), the National Eye Institute’s mission is to “conduct and support research, training, health information dissemination, and other programs with respect to blinding eye diseases, visual disorders, mechanisms of visual function, preservation of sight, and the special health problems and requirements of the blind.” The NEI was established in 1968. It has a budget of approximately $675 million (FY2014).
What if they could gather a few researchers for a small problem solving session? The objectives of this meeting would be the following:
i) To lay down step by step, what needs to happen for vision to be restored in a case of ND
ii) To identify from step (i), those areas where research is currently going on & those which are not the subject of current research
iii) To identify from step (ii), those areas where some ND specific research needs to be done in conjunction with ongoing research
If funds need to be arranged for this brainstorming to happen, then I would be happy to try and secure them. I don’t know how, but I’ll cross that bridge when I come to it.
My point is this: It is true that I can’t put a million dollars on the table for ND related research, but if we could define the way to solve the problem of vision (or lack of), then maybe I could find a way to obtain funds for individual projects.
So I went ahead and hammered out a letter to this effect.
Then I pondered for a bit about who to address it to. I figured I would send it to Dr. Paul Sieving, Director, NEI.
Then I pondered a bit more – what if the Norrie Disease Association (NDA) has had some dealings with the NEI in the past? In that case maybe they could tell me who to get in touch with. So I wrote to John Miller, President, NDA. No, the NDA has not been in touch with the NEI.
And yet I hesitated. I really want this brainstorming to happen. No more no’s. Perhaps I ought to route my request through the NDA? Perhaps that would strengthen my case? Hmmmm. Yes, I think I will try the NDA route. Let’s see where it leads.
Till next time then,
Meenu.